zAMP: Amplicon-based Metagenomics Pipeline for reproducible and scalable Microbiota Profiling
Protocol provided by Sedreh Nassirnia.
Microbiota profiling is a versatile and powerful tool with a wide range of applications in healthcare, research, environmental studies, agriculture, and industry. 16S amplicon sequencing helps us to understand the composition and functionality of microbial communities across various contexts, and can identify specific microbial patterns or biomarkers. For more on 16S sequencing see Amplicon Sequencing.
Applications
Disease diagnosis and prognosis: By profiling the microbiome, researchers can identify microbial signatures that are associated with certain diseases. These biomarkers can be used to diagnose or predict the course of a disease, enhancing the understanding and management of health conditions.
Treatment response prediction: Certain microbial taxa within the microbiome can serve as indicators of how a patient might respond to a particular treatment. This aspect of microbiota profiling is particularly valuable in personalizing medical treatments and the development of new therapeutic strategies.
Health and disease research: Beyond clinical applications, microbiota profiling is instrumental in broadening the scientific understanding of the role of the microbiome in health and disease. This research can lead to new insights into disease mechanisms and the development of novel preventive strategies.
Environmental and ecological studies: Microbiota profiling is not limited to human health. It’s also used in environmental and ecological research to understand the role of microbial communities in various ecosystems, contributing to conservation efforts and the study of biodiversity.
Agricultural and industrial applications: In agriculture, microbiota profiling can help to improve soil health, plant growth, and disease resistance. It can aid in processes like waste treatment and bioenergy production in industrial contexts.
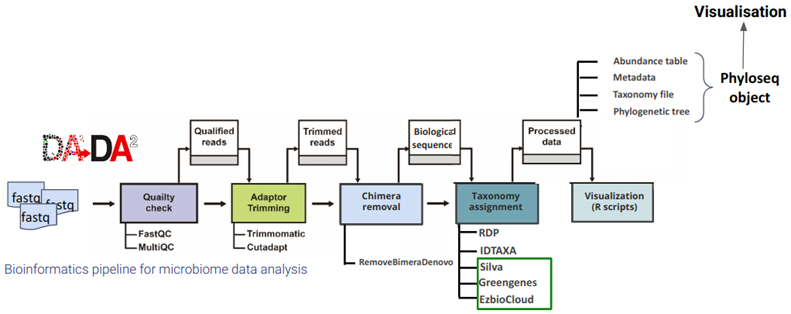
To effectively carry out microbiota profiling for these diverse purposes, we need standard protocols and tools that are compatible with many types of samples and able to provide accurate answers to the very diverse questions of the NCCR community. Therefore, we developed zAMP, an in-house customized, standardized, and automated DADA2-based data analysis pipeline, which includes a wide variety of command-line tools and R packages.
Implementation of zAMP
zAMP offers flexibility, allowing users to customize it depending on the research questions and sample type. To ensure reproducibility, zAMP integrates command-line tools and R packages as well as their dependencies through the Singularity container and allowed users to run and share microbiota profiling workflow.
Input Files for zAMP
The workflow accepts absolute paths to fastq files as input, paired-end sequencing reads from local storage, and raw reads from the Sequence Reads Archive (SRA), which will be downloaded with the SRA Toolkit.
Some of the Features
Read classification and taxonomy assignment: After the preprocessing steps, like merging paired reads, zAMP classifies reads using the assignTaxonomy function across multiple taxonomic ranks, from Kingdom to Species. This is facilitated by integrated classifiers like the original RDP, RDP as integrated into QIIME, and Decipher IDTAXA, all requiring a reference taxonomic database.
Reference taxonomic database preprocessing: The tool includes a secondary Snakemake workflow for preparing various reference taxonomic databases, such as Greengenes, SILVA, and EzBioCloud, enhancing the accuracy of taxonomic classification.
Dedicated scripts for taxonomic clarity: To avoid confusion between similar taxa, zAMP features a specific script that identifies and fuses taxa with identical sequences, providing clearer taxonomic information to the user.
In silico validation tool: Embedded within zAMP, this tool allows for accurate identification and classification of specific taxa or pathogens in clinical samples. Using a user-provided list of accession numbers for specific taxa, zAMP downloads genomes of interest from NCBI, performs in silico PCR using Simulate, and classifies amplicons using EzBioCloud or other user-specified databases.
Normalization of microbiome data: The pipeline offers various normalization methods, including rarefying, TSS, CLR, CSS, TM, and transformations like log, Logit, and Arc-Sine Square Root, ensuring robust downstream analysis.
Diverse output formats: zAMP generates outputs in multiple formats, including tab-delimited ASV tables, melted phyloseq table, and BIOM files. These are also combined into a single phyloseq object for easy manipulation and visualization in R.
Detailed visualization and statistics: Users benefit from a statistic table and barplot for raw, processed, and taxonomically filtered reads. The interactive Krona plot allows for detailed investigation of sample compositions, while rarefaction curves offer insights into species richness.
User-friendly execution: Designed for convenience, zAMP can be executed with a single command line, making it accessible for users to transform their microbiome data into meaningful insights.
For additional details about the tool and guidance on installation and tuning the parameters in the config file, please refer to the zAMP documentation and the GitHub repository.